苯丙酮尿症(Phenylketonuria)简称PKU, 是一种少见的遗传性代谢障碍疾病,是由于肝脏缺乏苯丙氨酸羟化酶所引起的代谢障碍。患儿主要表现为智力障碍、毛发变黄、皮肤色白及四肢短小,同时身上有一种特殊的霉臭气,尿有一种鼠臭味,有时有癫痫发作。
大家都知道,氨基酸对于人体来说,非常重要。其中有八种氨基酸是人体不能自身合成,也不能由其他途径转化,必须从食物中才能摄取,称为人体"必需氨基酸"。苯丙氨酸就是其中之一。当苯丙氨酸经食物中摄取后,部分为机体蛋白合成所利用,其余部分要经过肝脏,撒由其中的苯丙氨酸羟化酶转化为酪氨酸。而酪氨酸可进一步转化为多巴,肾上腺素,黑色素等等重要的生理活性物质。由于苯丙酮尿症患者肝脏缺乏上述的酶,一方面,苯丙氨酸不能转化为酪氨酸,从而在体内异常蓄积;另一方面,酪氨酸及多巴,肾上腺素,黑色素等等重要的生理活性物质的生成也受阻。这两个方面均会造成脑的损害,以及其他一系列的病理变化。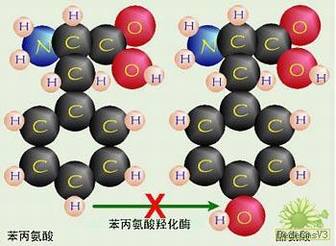
本症的主要危害为脑损害。未经治疗患儿在生后数月就会出现不同程度的智力发育落后,大多数有烦躁,抑郁等精神症状,最终将造成中度至极重度智力低下,成为家庭与社会的沉重负担。由于黑色素的缺乏,患儿表现头发较黄,皮肤和虹膜颜色浅。由于血中蓄积的苯丙氨酸的旁路代谢,产物经尿排出,常有一种令人极不愉快的鼠尿味;此外,患儿常合并湿疹,呕吐等。
值得重视的是,患儿在新生儿期和婴儿早期多无任何异常,症状随生长发育逐渐出现,并且多为不可逆转的脑损伤,而此时就医,往往会诊断为脑瘫或智力低能儿,一个家庭就背上了沉重的包袱。因此,只有通过新生儿筛查早期发现,早期治疗,才能保证疗效。
苯丙酮尿症在产前无法进行诊断,但出生后数天就能从血、尿中化验出来。所以目前很多国家已将这种病列为新生儿期普查的疾病。
最简单的诊断方法是"尿布试验",就是把几滴三氯化铁试剂滴在新生儿刚撒过尿的白布上,如果尿布呈现蓝绿色,就说明这个新生儿是苯丙酮尿症患者。也可以抽取新生儿血液,测定其中苯丙氨酸的含量。这种方法结果可靠,但不如尿布试验简便、安全。
诊断为苯丙酮尿症的新生儿,应立即开始治疗,即限制新生儿的食物中苯丙氨酸含量。据报道,生后立即治疗者,患儿即能正常发育,智力基本正常。如果等到五、六岁,小儿的大脑已经发生损害,再治疗就难以收到效果了。为此,每个新生儿都应该作尿布试验,以便早确诊,早治疗。对婴儿可喂给特制的低苯丙氨酸奶粉;为幼儿添加辅食时应有尽有以淀粉类、蔬菜和水果等低蛋白质食物为主。由于苯丙氨酸是合成蛋白质的必需氨基酸,缺乏时亦会导致神经系统损害,故仍应按每日30~50mg/kg适量供给,以能维持血中苯丙氨酸浓度在2~10mg/dl为宜。饮食控制至少需持续到青春期以后。